Cistiskā fibroze
Sinonīmi plašākā nozīmē
Cistiskā fibroze, plaušas
Angļu valodā: mukoviscidoze, cistiskā fibroze
Cistiskās fibrozes definīcija
Cistiskā fibroze ir iedzimta slimība. Mantojumu medicīniski dēvē par autosomāli recesīvu. Cistiskā fibroze (cistiskā fibroze) nav iedzimta X un Y dzimuma hromosomās, bet autosomālajā 7. hromosomā.
Lasiet mūsu vispārīgo rakstu par vielmaiņas traucējumiem: Vielmaiņas traucējumi - ko tas nozīmē?
Mutācija notiek uz tā saukto CFTR gēnu. Recesīvs nozīmēja, ka, lai slimība izdalītos, bija jābūt divām nepilnīgām gēna kopijām. Ja cilvēkam ir veselīga un mutēta gēna atrašanās vieta attiecīgajā 7. hromosomā, slimība nerodas.
Rezultāts ir patoloģisks gēna produkts. Tādējādi kodēts Hlorīda kanāli ir salauzti. Bojāti hlorīda kanāli noved pie biezu gļotu veidošanās visos eksokrīnajos dziedzeros.
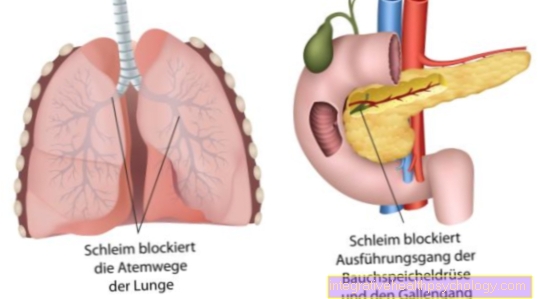
Šie eksokrīnie dziedzeri, t.i., dziedzeri, kas izdala sekrēciju uz ārpusi, ietver:
- aizkuņģa dziedzeris
- tievās zarnas
- elpceļu sistēma ar plaušām un bronhu sistēmu
- žults ceļu un
- arī Sviedru dziedzeri
Kopsavilkums
Cistiskā fibroze ir Iedzimta slimība. Tas tiek mantots tādā veidā, ka tas ir neatkarīgs no dzimuma un tikai ar divi bojāti gēni notiek. Tas ir visbiežāk sastopamā autosomāli recesīvā mantošana.
Sekas ir grūts gļotu veidojums visu eksokrīno dziedzeru, piemēram, plaušu, aizkuņģa dziedzera un arī sviedru dziedzeru. Tie ir balstīti uz to traucēta hlorīda transportēšana starp kameras iekšpusi un ārpusi (lasīt tālāk: Hlorīds asinīs). Mutācijas gēns ir ieslēgts 7. hromosoma un izraisa plašu orgānu iesaistīšanos ar attiecīgu iedarbību uz elpošanu, gremošanu un reprodukciju.
Diemžēl terapija var tikai mazināt simptomus, bet ne izārstēt. Dzīves ilgums pacientiem ar cistisko fibrozi salīdzinoši zems.
Tā kā tā ir recesīva iedzimta slimība, ir cilvēki, kas nes mainīto gēnu, bet necieš no pašas slimības. Šādas personas sauc Funkciju nesējs vai Diriģenti, t.i., pārvadātāji. Šiem cilvēkiem nav cistiskās fibrozes, jo otra gēna kopija ir neskarta, un slimā nav pietiekami spēcīga, lai dominētu.
Tomēr viņa var nodot šo nepilnīgo gēna kopiju saviem pēcnācējiem. Ja modificēts gēns jau būtu pietiekams, lai izraisītu slimību, tā būtu tā saucamā dominējošā mantošana. Šādu mantojumu var atrast, piemēram, Horeja Huntingtona. Jūs varat uzzināt vairāk par šo slimību zem mūsu tēmas Horeja Huntingtona.
Pie aptuveni 1:2500 atrodas Slimības biežums jaundzimušajiem Vācijā. Pārvadātājs ir par visiem 25. Vācijas iedzīvotāju skaitā.
galvenais cēlonis

Cistisko fibrozi izraisa gēna mutācija 7. hromosomā. Šī hromosoma ir autosomāla hromosoma, nevis dzimuma hromosoma.
Ikvienam ir 44 autosomālas hromosomas (katrai ir divas identiskas versijas) un divas dzimuma hromosomas. Šī 7. hromosomas mutācija noved pie bojātu hlorīdu kanālu veidošanās. Hlorīda reabsorbcija (atkārtota absorbcija) no dziedzeru sekrēcijām nav iespējama, jo receptori, kas ir hlorīda piesaistes punkts, nav iebūvēti dziedzera kanālos.
Tā vietā to nepareizi izskata un struktūras dēļ ieliek ieguvei. Tiek traucēta dabiskā hlorīda apmaiņa caur noteiktiem hlorīda kanāliem. Šos ts kanālus veido olbaltumvielas. Uz mūsu DNS tiek kodēts ļoti plašs olbaltumvielu klāsts. Hlorīdu kanālu ģenētiskā defekta dēļ no visiem dziedzeriem rodas dehidrētas un smagas gļotas, kas izdala to sekrēciju uz ārpusi. Tad gļotas daļēji bloķē kanālus vai plaušās elpceļus.
Lasiet arī par šo Hromosomu mutācija
Cistiskās fibrozes diagnostika

Tipiski simptomi, kas sākas zīdaiņa vecumā, ir revolucionāri cistiskās fibrozes diagnoze.
Šīs aizdomas pastiprina pozitīvā ģimenes anamnēze (tēva / mātes vai tuvu radinieku slimība). Pozitīva ģimenes anamnēze nozīmē, ka ģimenē ir vai jau ir bijuši cistiskās fibrozes gadījumi - mātes vai tēva pusē.
Aizkuņģa dziedzera enzīmu trūkumu var noteikt arī izkārnījumos. Jebkādus elpceļu aizsprostojumus var noteikt, rentgena starojot krūtīm.
Cistiskās fibrozes diagnostikā palīdz arī sviedru pārbaude, kurā mēra hlorīdu saturu sviedros. Ja tiek pārsniegta noteikta vērtība un piemērojami arī citi simptomi, diagnoze ir salīdzinoši noteikta. Bieži vien paši vecāki pamana paaugstinātu sāls saturu zīdaiņa sviedros.
Nedzimušu bērnu var pārbaudīt arī attiecībā uz šo iedzimto slimību. Izmantojot amnija šķidruma punkciju (Amniocentēze) augļa šūnas noņem un pārbauda, vai nav mutācijas gēna.
Lasiet vairāk par tēmu: Bērna rentgena pārbaude
Cistiskās fibrozes terapija
Ikviens, kuru skar cistiskā fibroze, saņems padomu vienā Cistiskā fibroze - ambulatorā nodaļa vai padoms no Cilvēka ģenētiķis (Speciālists iedzimtu slimību gadījumā) ieteicams. Tie var palīdzēt uzlabot dzīves kvalitāti vai, ja vēlaties bērnus, aprēķiniet slima bērna varbūtību. Ja vecāki ir auglīgi un auglīgi.
Pretējā gadījumā ārstēšana ir simptomātiska, jo cēloni - nepilnīgu gēnu - nevar novērst.
Neārstējama slimība
Cistiskā fibroze (cistiskā fibroze) joprojām ir neārstējama slimība.
Cistiskās fibrozes gadījumā ir svarīgi, lai būtu pietiekami daudz sālsNātrija hlorīds, NaCl). Mukolīze, protams, ir vērsta. Mukolīze ir gļotu, īpaši plaušās, izšķīšana, lai atvieglotu elpošanu.
Zāles un ieelpošana var mazināt simptomus. Ja plaušu darbība ievērojami pasliktinās, var dot skābekli.
Ar intensīvas fizioterapijas palīdzību (fizioterapija), piemēram, pieskaroties masāžai un elpošanas vingrinājumiem, ārstē arī cistiskās fibrozes izraisītās plaušu izmaiņas.
Bieži slimība beidzas ar nepieciešamo plaušu transplantāciju. Tomēr gaidīšanas saraksti ir gari.
Perorāla aizkuņģa dziedzera enzīmu un taukos šķīstošu vitamīnu ievadīšana ir arī terapijas sastāvdaļa. Tāpēc aizkuņģa dziedzera uzdevums ir jāatbalsta vai drīzāk jāaizstāj. Taukos šķīstošie vitamīni ir A, D, E un K. Tie jāievada tieši asinīs, jo gremošanas enzīmu trūkuma dēļ tos nevar absorbēt no pārtikas.
Uzturā vajadzētu būt arī daudz kaloriju, jo tikai daļu no tām var iegūt no pārtikas.
Lai izvairītos no papildu riska faktoriem tādām komplikācijām kā gripa vai pneimonija, bērns jāvakcinē. Ieteicams veikt šādas vakcinācijas:
- masalas
- Pneimokoki
- gripa
Lasiet vairāk par tēmu: Superinfekcija
Protams, šiem pasākumiem nepieciešama konsultācija ar ārstu, ar kuru jāapspriež riski.
Mūsdienās ģenētiskajā izpētē tiek liktas lielas cerības uz cistiskās fibrozes terapiju. Trūkst ģenētiskās informācijas tiek mēģināts ieviest cilvēka genomā. Mēs meklējam vektorus, kas varētu apgūt šo uzdevumu. Vektoriem var būt, piemēram, baktēriju vai vīrusu DNS, kuriem izdodas iekļaut veselīgu biežumu mūsu ģenētiskajā veidojumā.
Pašlaik tiek pārbaudīta terapeitiskā pieeja nedzimušiem pacientiem. Pelēm peļu embrijiem jau ir izdevies ar amniocentēzes (amnija šķidruma inokulācijas) palīdzību ieviest veselīgo gēnu, kurā bija pareiza gēna secība. Tādējādi šīm pelēm tika iegūts veselīgais CFTR gēns. Amniocentēze ir bērna šūnu punkcija un izņemšana no amnija šķidruma.To veic caur mātes vēdera sieniņu.
Tomēr Vācijā šī intrauterīnās (= dzemdē = dzemdē) "terapijas" forma ir aizliegta.
profilakse
A preventīvs pasākums šajā ziņā tas neeksistē, jo tā ir iedzimta slimība.
Tomēr var apmeklēt cilvēku ģenētisko konsultāciju centru (parasti atrodams universitāšu slimnīcās). Šeit tiek aprēķināts, cik liels risks būtu slimības nodošana bērniem.
Šis padoms vienmēr ir noderīgs, ja ģimenes anamnēzē ir cistiskā fibroze.
Arī vienu Pirmsdzemdību diagnostika ir vērts tiekties. Šeit pirms dzimšanas (t.i., pirmsdzemdību periodā) a Amnija šķidruma pārbaude (Amniocentēze) Izpildīts. No amnija šķidruma tiek ņemtas augļa šūnas (bērna šūnas), un DNS tiek pārbaudīts, vai nav mutācijas gēna.
Cistiskās fibrozes prognoze
Diemžēl vidējais dzīves ilgums pacientiem ar cistisko fibrozi ir tikai 32-37 gadi. Mūsdienās tiek lēsts, ka jaundzimušo dzīves ilgums, kas dzimuši ar šo stāvokli, ir aptuveni 45-50 gadi.
Prognoze ir ļoti atkarīga no terapijas un no tā, vai tā tiek ievērota.
Tāpēc svarīgu lomu spēlē pats pacients un viņa motivācija.